
What are prions?
This post is part of a series introducing the basics of prion disease. Read the full series here.
The term prion was coined to mean proteinaceous infectious particle [Prusiner 1982]. It’s usually pronounced PREE-on in the U.S. and PRY-on in the U.K. Prions are bits of misfolded protein that have the ability to spread by making other proteins misfold.
The concept and the nomenclature surrounding it are easier to understand with a bit of historical background.
A couple of centuries ago, people started taking note of a unique disease afflicting sheep, which they called scrapie because the sheep became deranged and, among other things, started scraping their hindquarters raw. Scrapie was an unusual disease: it seemed to spread like a virus but in other ways it looked like a genetic disease [Parry 1962]. Most peculiar of all, the infectious agent – whatever it was – seemed nearly indestructible. Viruses can usually be inactivated by heat and then used in vaccines, but scrapie survives high temperatures. In one notable incident, a herd of sheep were vaccinated against louping-ill virus with a heat-treated sample from other sheep [Gordon 1946]. The sample turned out to contain scrapie, and many of the inoculated sheep became ill with scrapie as a result.
People studied scrapie for about 150 years without much insight as to the true nature of the disease. A breakthrough came in the mid-20th century when scientists started studying kuru, a neurological disease epidemic in the highlands of Papua New Guinea. Carleton Gajdusek won the 1976 Nobel Prize for demonstrating that kuru could be transmitted to chimpanzees [Gajdusek 1966], as could Creutzfeldt-Jakob disease (CJD), another condition which wreaked havoc on people’s brains. The damage from CJD looked a lot like that of kuru and scrapie under the microscope.
Soon the nature of the infectious agent became clear. The “scrapie agent,” as it used to be called, was a protein [Prusiner 1982]. Unlike bacteria and viruses, it has no DNA or RNA. This was a new thing in all of biology, and Stanley Prusiner won the 1997 Nobel Prize for demonstrating this [Prusiner 1998].
So first there was the “scrapie agent”, and then this was shown to be an infectious protein which Prusiner dubbed a “prion”. Then when Prusiner discovered the protein of which prions were made, he called it “prion protein” (PrP for short) [Prusiner 1983]. Next it turned out that PrP was a protein that everyone – even people who aren’t sick – have in their bodies, and that there’s a gene in your DNA, on chromosome 20, that contains instructions telling each cell how to make PrP [Oesch 1985]. This gene was eventually called the PRNP (prion protein) gene.
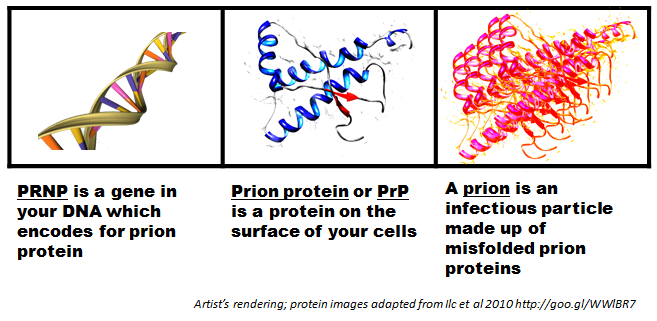
If everyone has prion protein, then why do most people never get sick with a prion disease? It turns out that PrP normally exists in a healthy state called “cellular prion protein” or PrPC. But it’s capable of misfolding into a “scrapie prion protein” or PrPSc. One particle of PrPSc can cause other PrPC to convert into PrPSc.
In the novel Cat’s Cradle by Kurt Vonnegut, there’s a sort of doomsday weapon called ice-nine. Ice-nine is a fictional water crystal that has a higher melting point than ordinary ice – it’s solid at room temperature. One crystal of ice-nine causes other water molecules to become ice-nine and so it can freeze an entire ocean. PrPSc is like ice-nine: it “teaches” the other prion proteins how to fold up into a disease state [Kocisko 1994].

When Stanley Prusiner coined the term prion, there weren’t many known prion diseases – just kuru, scrapie, and Creutzfeldt-Jakob disease, all of which turned out to be caused by the same protein, PrP. We now know that several other diseases – mad cow or bovine spongiform encephalopathy in cows, chronic wasting disease in deer, and fatal familial insomnia and Gerstmann-Straussler-Scheinker syndrome (just call it GSS!) in humans are all prion diseases, also caused by PrP.
And in fact, it turns out that the idea of proteins that can “teach” other proteins how to fold isn’t limited to just PrP. There is now a growing body of scientific evidence to suggest that other neurodegenerative diseases such as Alzheimer’s and Parkinson’s disease are also caused by different proteins that can act like ice-nine [Stohr 2012, Luk 2012, Prusiner 2012, Jucker & Walker 2013]. Even more amazingly, it turns out that the idea of proteins that cause other proteins to fold up like themselves isn’t always a bad thing. In yeast, it appears to be part of every day life – yeast cells will divide and pass on their “prions” to their daughter cells kind of like they pass on DNA, and those proteins will contain important information that could help the daughter cells survive [Halfmann & Jarosz 2012].
So over the past several years, it’s increasingly common to hear people use the term “prion” to refer to amyloid beta (the protein involved in Alzheimer’s), alpha synuclein (the culprit in Parkinson’s), and various proteins in yeast. When we say “prion disease” here at Prion Alliance, we’re almost always referring just to the “original” prions – the misfolded PrP particles that cause Creutzfeldt-Jakob disease, fatal familial insomnia, and so on. Those are the diseases that we’ve made it our mission to cure. If you want to be very precise about terminology, you can call these “PrP prion diseases.”
Want to learn more? For a fascinating history of prions and prion diseases, we recommend The Family That Couldn’t Sleep by D.T. Max (2006).