
What are the potential treatments for prion disease?
This post is part of a series introducing the basics of prion disease. Read the full series here.
If you’ve read the other entries in our FAQ you know that the human prion diseases – Creutzfeldt-Jakob disease (CJD), fatal familial insomnia (FFI), and Gerstmann-Straussler-Scheinker syndrome (GSS) – are all caused by prions made of the same protein, called prion protein or PrP. Prion diseases can come in genetic, sporadic or acquired forms. The bad news is that as of today, prion diseases are completely incurable and untreatable. The good news is that we and many other groups worldwide are actively working to find a treatment or cure for these diseases.
Prion diseases are different from most other types of diseases. Prions can act like an infection as they travel from cell to cell in a person’s body, yet unlike viruses or bacteria, they have no DNA or RNA – they’re just made of protein. There has never before been a treatment for a disease like this, so right now it isn’t yet clear which strategy will ultimately lead to a treatment or cure. Scientists are investigating a lot of different possible ways of treating these diseases.
small molecules
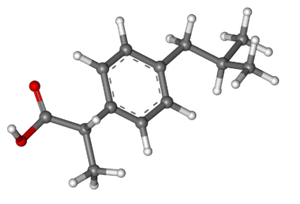
Odds are that most drugs you’ve ever taken, for anything from a headache to a bacterial infection to contraception, have been what we call small molecules. For instance, ibuprofen, the painkiller whose molecular structure is shown above, is a small molecule. This just means it is made of relatively few atoms – 33 atoms in the case of ibuprofen.
The “small” in small molecule is important because it means that these drugs are often (though not always) pretty easy to administer. Think about painkillers for a moment – when you swallow a pill, that molecule is able to get from your stomach to just about every part of your body, which is why it can work whether you’ve hurt your foot or your neck. This is a major advantage of small molecules that isn’t necessarily true of the other therapeutic approaches we’ll talk about in a minute.
Several research groups worldwide are working on finding small molecule drugs that can treat prion disease. For instance, efforts are actively ongoing at the Institute for Neurodegenerative Diseases at UC San Francisco, the MRC Prion Unit in London, the prion center at LMU in Munich, and the prion group at Scripps in Florida.
While none of these groups have found anything that’s ready for a clinical trial in humans just yet, they’ve made several promising steps forward. In fact, scientists have already discovered a few small molecules which work really well against mouse prion strains – cpd-b, IND24, and anle138b [Kawasaki 2007, Wagner 2013, Berry 2013, Lu & Giles 2013]. The first two of those turned out not to work against common human prion strains; we are currently testing anle138b. Meanwhile, new drug discovery efforts are continuing. The biggest remaining challenge will be to find a molecule that will work against multiple human prion strains.
It’s worth mentioning that over time, several different existing drugs have also been proposed as possible treatments for prion disease – for instance, quinacrine and doxycycline. We don’t think the experimental evidence is very strong for any existing drug being an effective treatment for prion disease. We’ll review a few of the most often-discussed candidates in an upcoming FAQ, but for now, suffice it to say we expect that finding a treatment will require discovering a brand new drug and bringing it to market specifically to treat prion diseases.
antibodies
Antibodies are proteins produced by the immune system which can bind to a particular “enemy” target – usually another protein. Your body can produce antibodies naturally in response to viral infections, bacterial infections, cancer, and so on – they’re a central part how you fight off infections. And once your body has found an antibody against something bad, it remembers how to make it for future reference – that’s why people usually don’t get chicken pox twice.
Modern science is also capable of manufacturing antibodies outside the body to be used as treatments for diseases. If you’ve taken a drug other than a small molecule, odds are good that it was an antibody – some of the most common ones are Humira and Herceptin. Antibodies have to be injected, not taken orally, and they’re often for more serious conditions like autoimmune diseases or cancer – and not just for headaches.
Antibodies against PrP could be one potential treatment for prion diseases. Potential treatments for any disease usually start in a test tube, then move on to a mouse, and then finally move on to humans. Antibodies are no exception. When prion-infected cells in a dish were treated with antibodies against PrP, the infection was completely cleared [Enari 2001, Peretz 2001]. Shortly thereafter, antibodies were shown to be effective in treating prion-infected mice under certain conditions [White 2003]. As of earlier this year, the MRC Prion Unit in London has started discussing the possibility of launching a clinical trial of an anti-PrP antibody named PRN100 to treat sporadic Creutzfeldt-Jakob disease. As of this writing, no trial has officially been announced and the MRC Prion Unit has not provided any estimates of when a trial might begin.
The major challenge for such a clinical trial will be that antibodies are huge – around a thousand times larger than most small molecule drugs. One difficulty in developing drugs for prion diseases – or any other brain disease – is that in order to be effective, any treatment will need to get past the blood-brain barrier. The cells surrounding blood vessels in the brain are so tightly packed that even many small molecule drugs can’t get from the blood into the brain – only a special subset can do so. Antibodies are so large that only a vanishingly small amount of them can cross the blood brain barrier.
In the original mouse study that showed how antibodies could be effective, some groups of mice were infected with prions peripherally, meaning outside of the brain [White 2003]. When these mice were treated early on with antibodies, the antibodies were able to cure the infection before it ever reached the brain, and the mice never got sick. That’s an amazing result, and to this day, antibodies are the only treatment shown to completely cure peripheral prion infections in mice. However, when mice were infected directly in their brain or when they weren’t treated until after the infection had reached the brain, the antibodies had no detectable effect: the mice died at the same time as mice that weren’t treated with any antibodies.
As far as we know, sporadic and genetic prion diseases in humans start in the brain, so there’s no opportunity to treat them in the periphery. From what we know right now, a clinical trial, if one happens, would most likely involve intravenous injections of antibodies in patients who are already experiencing symptoms. This approach hasn’t worked in mice [White 2003], presumably because not much of the antibody got into the brain. The folks at MRC Prion Unit have pointed to evidence that the blood-brain barrier may be broken down in prion disease patients [Chung 1995, Brandner 1998], which would help the antibodies to get across, and as noted here they’ve also raised the possibility of eventually administering antibodies directly into the brain instead of into the bloodstream. In animal studies, direct injections of antibodies into the brain have been shown to have some small effect (survival was extended by ~8%) though the animals were not cured [Song 2008 (ft)].
Overall, evidence in the scientific literature suggests that antibodies will probably not be a cure or really dramatically extend life in prion disease patients who are already experiencing symptoms, but they may prove to have some helpful effect. Moreover, the improved delivery of antibodies to the brain is an area of active research (see for instance the recent Brain Shuttle announcement). So even if antibodies don’t work on the first try, they may be a really valuable treatment once a better delivery system comes along.
gene silencing

Gene silencing is the use of RNA or DNA to “turn off” a gene as a treatment for a disease. This idea is much more experimental – many more years away from clinical trials – than small molecule drugs or antibodies – but in the long run, it might prove very promising.
One thing we know for sure about prions is that you can’t have a prion disease without PrP [Bueler 1993]. When mice have been infected with prions and then later the prion protein gene was turned off, the mice fully recovered from the disease [Mallucci 2003]. So if we could figure out a way to turn off the prion protein gene, PRNP, in humans, and make it stop producing PrP, then we might be able to cure prion diseases altogether, even after patients have gotten sick.
But that’s a big if. When researchers shut off the prion protein gene in mice, they used a special genetic manipulation that isn’t possible in humans. Now, some researchers are looking for small molecule drugs that can make the body produce less PrP [Karapetyan & Sferrazza 2013, Silber 2014], but no really strong candidates have emerged just yet. So another possibility is to use gene silencing to shut off the supply of PrP. There are two major technologies for gene silencing that you’ll hear talked about – RNA interference (RNAi) and antisense oligonucleotides (ASOs) - but the core principles of the two are similar. Remember that the prion protein gene in your DNA has a message that gets converted into RNA, and then the message in the RNA gets translated into protein. Gene silencing means using tiny pieces of RNA, DNA, or chemically modified versions of those, that stick to the prion protein RNA and can either keep that RNA from being translated into protein, or can cause the cell to destroy the RNA altogether.
In principle, gene silencing is a brilliant idea, but it has some serious problems we haven’t worked out yet. First and foremost, it has the same problem as antibodies: the pieces of RNA or DNA used are much, much larger than small molecules. They cannot cross the blood-brain barrier and they don’t move around much within the brain either. When scientists have injected them into the brains of prion-infected mice, they’ve only been able to reach a small area of the brain, and they only saw a small therapeutic effect, extending survival by about 18% [White 2008]. When you consider how much larger a human brain is than a mouse brain, odds are we couldn’t see much of an effect at all with today’s technology.
But the good news is, loads of scientists worldwide are working on figuring out how to deliver gene therapy to the brain and other parts of the body. Most of them are working on other diseases and not on prion disease in particular, but whatever techniques they come up with will probably be applicable to prion disease too. As of today, gene therapy for prion disease is still a little bit science fiction, but there’s a chance that it could be a really effective therapy in 10 or 20 years. We think gene therapy is well worth investing our research energy in for the potential long-term benefit – but don’t expect to be talking to your doctor about it anytime in the next few years.
a quick note about other technologies
Above we’ve introduced the three types of treatments we think could potentially be promising for prion disease: small molecules, antibodies, and gene silencing. In the interest of thoroughness, we should also briefly address a couple of other treatment strategies.
One sometimes hear talk of vaccines for prion disease. Because prion diseases can in rare cases be acquired as infections, it is tempting to make analogies to viruses, and to the huge advances in public health that have been made through vaccination campaigns against viruses like smallpox and polio. But there are many reasons to think this strategy won’t work for prion disease. PrP isn’t a foreign pathogen, it’s a protein your own body produces, so causing the body to mount an immune response against PrP is both difficult to do and potentially dangerous. There are researchers working on strategies to address those problems, but there’s also a much bigger issue which no research has yet addressed. As far as we know, most prion disease starts in the brain, and the immune system doesn’t normally attack proteins in the brain. Adding to that, patients almost never know they have prion disease until after they already have symptoms, but vaccinations are by nature a pre-emptive treatment – they need to be given before an infection starts. Anti-prion vaccine research has not addressed these fundamental limitations of vaccine technology. For this reasons we do not expect that vaccines will be a part of the treatment strategy for prion diseases.
Another proposed strategy is stem cell therapy for prion disease. This has been proposed in various forms: it could involve trying to generate new brain cells to replace ones that patients have lost, or it could involve adding extra, genetically modified cells to a patient’s brain to produce special proteins or other therapeutic agents that will slow down prion infection. Our view is that any sort of stem cell therapy is probably quite a long way from being useful to prion disease patients. That’s not to say it couldn’t happen someday – it is an active area of research for Parkinson’s disease, for instance, and we’ll see what new technologies are developed.
If you made it this far, your next question may be: when will these new treatments become available? Read our next post to learn more.